Update
The web based Sample Scheduler has been replaced with a downloadable Java Application called ScheduleTool.
To install the program follow the installation instructions below in the documentation.
NUS ScheduleTool Documentation
The ScheduleTool was created to assist users in the creation of Non-Uniform Sampling (NUS) schedules for the collection of NMR data. The tool was designed with ease of use in mind. By entering easily know values for molecular weight, nucleus types, number of dimensions, and sweep widths the program will automatically determine suitable values for parameters directly responsible for building the random sample schedules. A graphical user interface was built to aid in this process and to visualize the sample schedules. Analysis tools are also built into the tool for developers and to help educate users as to why some sample schedules may be better than others.
While the ScheduleTool was designed with ease of use in mind it also supports more advanced features and the ability to run from the command line for advanced users and developers who may be generating many sample schedules for testing purposes rather than a single sample schedule for NMR data collection.
While I attempted to think of as many features as possible I am sure there are many more features users may want to see added. Send any requests for new features, report bugs, or questions about the tool to markm@neuron.uchc.edu.
The ScheduleTool was created by Mark Maciejewski and Val Gorbatyuk in conjunction with Jeffrey Hoch at the University of Connecticut Health Center. Thanks to Alan Stern, Mehdi Mobli, and Jay Vyas for helpful discussions.
OUTLINE
INSTALLATION (top)
Note: A semi recent version of java must be installed to run the Schedule Tool (java version 1.5 or higher). Use the command "java -version" from a command line to determine the version installed on your system.
Download the latest version of the ScheduleTool (ScheduleTool-dir.tar), unpack, and save the jar file and ScheduleTool script to a location in the system path. NOTE: OSX installations should use the ScheduleTool-mac script. The script simply runs the command:
java -jar -Xmx1024m ScheduleTool-date.jar $@ Note: The date part of the filename for the ScheduleTool will change as the program is updated. The OSX version has an additional argument which places the name of the program on the dock when it is running.
The -Xmx1024m allows the program to use up to 1GB of memory. This number can be adjusted based on your needs. Note that if you encounter program errors, especially for very large 3D sample schedules, you may want to try increasing the 1024 to a larger value, say 2048. The $@ passes all arguments on the command line to the program.
KNOWN BUGS or are they FEATURES? (top)
- Problems can arise with saving sample schedules along with the results from the peak picker when more than a single sample schedule window is open at a time. It is strongly advised to create a single sample schedule, review, analyze, and save it, and then close the sample schedule window before creating another sample schedule. The main ScheduleTool program may remain open between creating additional sample schedules, just make sure that there is only a single schedule window open at a time.
- When creating sample schedules from the command line the molecular weight and nucleus type(s) must be entered. While these values are not used directly to create the sample schedule, they are used to automatically calculate reasonable values for parameters used to create the sample schedule and for some of the statistics that are reported. Any parameters entered on the command line will overwrite these default values calculated from the molecular weight and nucleus type(s). Thus if all the parameters needed to create a sample schedule directly are enetered on the command line, and you don't care about the statistics, you may feel that it is unnecssary to enter the molecular weight and nucleus type(s). While I agree with this, the tool does not allow the molecular weight and nucleus type(s) to be skipped. I may build the logic into the program to allow these to be eliminated in the future.
- Command line parser. The command line parser is not very smart. It reads required values one at a time until it reaches an un-required value. At that point default values for various parameters are determined. It then continues to read values from the command line and overwrites any default values determined after the default values were read. Thus it is critical that when using the command line that the default values are entered first and the non required values entered at the end.
NON-UNIFORM SAMPLING (NUS) (top)
A description of non-uniform sampling can be found at http://rnmrtk.uchc.edu/rnmrtk/NUS.html
Non-uniform sampling is the process of collecting time-domain data at non-fixed intervals. There are several advantages to collecting data in this manner.
- Data sets can be collected with fewer points and hence can be quicker to acquire.
- Sample schedules can be created that increase the sensitivity per unit time. While a sample schedule collected with only a subset of the points as compared to a uniformly sampled spectrum will have a lower overall sensitivity (assuming that the uniformly sampled time points do not exceed ~1.26X T2), the decrease in sensitivity is often small.
- For example a sample schedule with 10% of the possible points collected may have a sensitivity which is 65% that of the uniformly sampled spectrum and it is collected 10X faster.
- NUS sample schedules can be created that increase resolution, especially for high field instruments.
- Spectroscopists are often limited, due to time, on the total number of increments that they can acquire in indirect dimensions. In the direct dimension spectroscopists often collect data out to 3X T2 to maximize resolution. However, for indirect dimensions they often only collect out to a small fraction of T2 as that is all that time allows.
- This problem is exasherbated at higher field strengths where sweep widths are larger and thus dwell times shorter. Thus the higher potential resolution afforded with higher field instruments is rarely achieved due to time constraints that restrict sampling along the indirect dimensions to large enough evolution delays to achieve the desired resolution.
- NUS eliminates this constraint as the percentage of points that need to be collected is small so that the maximum increment along the indirect dimensions can be set higher to achieve higher resolution.
ALGORITHM (top)
The algorithm for generating the sample schedule is based on randomly picking a subset of possible points from a 1D, 2D or 3D grid, but with a skewed random distribution based on an exponentially decaying function in each dimension. First the algorithm finds the maximum increment in all dimensions to determine the size of the 1D, 2D, or 3D grid of potential points that may be picked (this would be the size of a uniformly collected spectrum). The algorithm then uses the decay rate in each dimension along with the sweep width in each dimension (used to determine the evolution time of each potential point) to determine the probability of each potential point by:
1D: Probability = [EXP (t1*decayRate_t1)]
2D: Probability = [EXP (t1*decayRate_t1)] * [EXP (t2*decayRate_t2)]
3D: Probability = [EXP (t1*decayRate_t1)] * [EXP (t2*decayRate_t2)] * [EXP (t3*decayRate_t3)]
where tn = (increment_number * (sweep_width_tn)-1)
The initial point with an evolution time equal to zero has a probability set to 1 and other points have a probability between 0 and 1. Note that for constant time experiments the decay rate is set to zero for that dimension. Also note that for SIN or COS modulated signals the appropriate SIN or COS function is multiplied to the exponential decay function above to determine the probability of each point.
New Probability = Probability (above) * ABS [COS/SIN(3.14 * J_tn * tn)], where tn = (increment_number * (sweep_width_tn)-1)
Once the probability of each potential point is determined that probability is multiplied by a random number between 1 and 0. Thus a potential point with a high probability based on the decay rate may end up with a low probability if the random number is close to 0. Thus the product of the probability based on the decay rate and J-coupling along with the random number is what leads to the randomness of the process. Once each potential point has its new probability based on the product of the decay rate and random number the values are sorted from greatest to least. The sample schedule then consists of the highest values in the sorted list up to the total points in the sample schedule.
RUNNING THE SCHEDULE TOOL WITH A GRAPHICAL USER INTERFACE (GUI) (top)
Note: The ScheduleTool may be run with a Graphical User Interface (GUI) or standalone from the command line. Any command line arguments entered when running the program in GUI mode are passed to the program and those arguments are used to populate the program entries with default values. NOTE: The default values ndim, molWeight, field, swtN, and nucltN must be entered first. All other values must come after these values are entered.
- Running the program in generic GUI mode
- Enter the command "ScheduleTool" or "ScheduleTool --gui"
- Running the program in help mode which shows all arguments which may be passed to the program
- Enter the command "ScheduleTool --help"
- Running the program in GUI mode with default parameters passed to the program
- Example: "ScheduleTool --ndim 2 --molWeight 32000 --field 600 --swt1 8000 --swt2 1750 --nuclt1 13C --nuclt2 15N --gui"
- This command will invoke the GUI of the ScheduleTool and populate the number of dimensions to 2, the Molecular Weight to 32,000 Da, the sweep widths to 8000 and 1750 Hz and the nuclei to 13C and 15N. Those values will then be processed to determine reasonable values for Maximum Increment, Decay Rate, and Total Points and the determined values will be populated in the GUI text boxes upon opening the program, as if the Compute Defaults button were pressed. Any of the values may be changed by the user prior to creating a sample schedule.
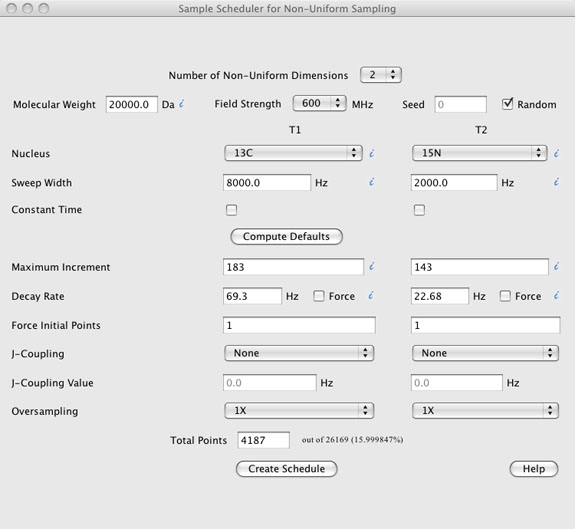
DESCRIPTION OF THE GRAPHICAL USER INTERFACE (top)
The GUI has seven components.
- The "Number of Non-Uniform Dimensions" located at the very top.
- This is the number of non-uniform sampled dimensions, not the number of dimensions in the experiment. The acquisition dimension is typically collected uniformly.
- Sample information including "Molecular Weight", "Field Strength", "Nucleus" type, "Sweep Widths", and whether the experimental dimension is "Constant Time". A checkbox for using a "Random" seed is also present along with a text box to enter a "Seed" value if the random checkbox is not checked.
- A "Compute Defaults" button. This button, when pressed, uses the sample information described above to compute default values for the "Maximum Increment" "Decay Rate", and "Total Points". Other values such as "Force Initial Points", J-Coupling", and "Oversampling" are not altered by the "Compute Defaults" button.
- Sample schedule information is located below the "Compute Defaults" button and includes text boxes for "Maximum Increment", "Decay Rate", "Force Initial Points", "J-Coupling Value", and "Total Points" along with pull down menus for "J-Coupling" type, and "Oversampling". The values in this sections, along with the "Sweep Widths" entered in the sample information section are used to compute the sample schedule.
- The "Create Schedule" button creates the sample schedule and opens a Sample Schedule window with the ability to view the sample schedule, view the point spread function (PSF) of the sample schedule, provides some basic statistical information about the sample schedule and parameters used to create it, and provides pull down menus for saving the sample schedule and other information. Each of these are described in more detail below in the sample schedule window help.
- The "Help" button is a web link to this document
- Information mouse over's are shown as blue italic i's.
 When the mouse hovers over any of these additional information is presented to the user. For example, when the mouse is placed over the "Molecular Weight" information mouse over, the user is presented with the molecular rotational correlation time that will be used to compute default T2 relaxation times based on the "Nucleus" choice when the "Compute Defaults" button is pressed.
When the mouse hovers over any of these additional information is presented to the user. For example, when the mouse is placed over the "Molecular Weight" information mouse over, the user is presented with the molecular rotational correlation time that will be used to compute default T2 relaxation times based on the "Nucleus" choice when the "Compute Defaults" button is pressed.
DETAILS OF GUI COMPONENTS (top)
- Number of Non-Uniform Dimensions
(top)
- Pull-down menu to choose the number of non-uniform dimensions to be used to create the sample schedule. Note that the acquisition dimension is normally collected uniformly so that the number of dimensions selected here is generally at least one less than the number of dimensions in the experiment. The GUI reformats automatically based on the number of dimensions to include a column for each dimension.
- Molecular Weight
(top)
- Enter the molecular weight of the protein to be studied. The molecular weight is used to compute a molecular rotational correlation time which is then used to estimate expected T2 relaxation times to compute default values for parameters to create the sample schedule.
- The information mouse-over shows the molecular rotational correlation time estimated for the entered molecular weight.
- The MW does not need to be entered properly to create the sample schedule and is only used by the "Compute Defaults" button to estimate appropriate values to create the sample schedule and some simple statistics determined after the sample schedule is created. If you are entering all the parameters needed to create the sample schedule by hand and do not care about the statistics then the MW value entered is irrelevant.
- Field Strength (top)
- The field strength can be selected from 500 to 900 MHz in 100 MHz intervals. The field strength is used when calculating T2 rates for 13CO and 15N nuclei as these nuclei are affected by CSA. Field strength is ignored for other nuclei as the it affects the T2 rate to a smaller extent.
- Seed
(top)
- Sample schedule points are selected randomly. The user can define a seed value if desired to ensure the exact sample schedule can be recreated (assuming all relevant parameters are the same).
- Random
(top)
- A checkbox that will randomly generate a seed value for the creation of the sample schedule.
- Nucleus
(top)
- Pull-down menus for each of the dimensions with choices for nuclei. The choice of nucleus along with the molecular weight and field strength are used to estimate expected T2 relaxation rates.
- The information mouse-over shows the estimated T2, R2, and linewidth for the given nucleus, field strength, and molecular weight.
- The nucleus does not need to be entered properly to create the sample schedule and is only used by the "Compute Defaults" button to estimate appropriate values to create the sample schedule and some simple statistics determined after the sample schedule is created. If you are entering all the parameters needed to create the sample schedule by hand and do not care about the statistics then the nucleus choices entered are irrelevant.
- Sweep Width
(top)
- The sweep widths for each of the dimensions are entered in Hz.
- Sweep widths are required to create the sample schedule as well as to compute default values for the "Maximum Increment" when the "Compute Defaults" button is pressed.
- The information mouse-over shows the dwell time for the given sweep width.
- Constant Time
(top)
- Checkbox if a given dimension is to be collected as a constant time dimension. If checked the "Decay Rate" text box is automatically set to 0.0 when creating the sample schedule and sample points are selected purely randomly without any skewing of the data points to early evolution times.
- Semi-constant time experiments should leave this checkbox UN-checked.
- Compute Defaults
(top)
- The compute defaults button uses sample information provided above the "Compute Defaults" button to populate the maximum increment, decay rate, and total points text boxes below the "Compute Defaults" button with values. The values are not necessarily optimal values, but should be reasonable.
- In detail the compute defaults button determines a rotational correlation time based on the molecular weight of the molecule.
- The rotational correlation time is determined based on a plot of MW versus correlation time from several 15N relaxation studies in the literature rather than a theoretical value. This value is slightly higher than the theoretical value.
- Based on the rotational correlation time and the nucleus choice, estimates for T2 relaxation rates are determined.
- These are based on plots of experimental T2 rates for different nuclei versus correlation time and are not theoretical.
- Based on the estimated T2 values and the sweep widths, a default value for maximum increment is determined which leads to the maximum dwell time of around 1.26X T2.
- Note that maximum resolution is achieved at around 3X T2 and that any data points collected beyond 1.26X T2 will lead to a larger increase in noise than signal although help resolution up to around 3X T2.
- In addition to the maximum increment the estimated T2 rates are also used to determine a default value for decay rate.
- By default the decay rate is set to 1.26X that of R2 for a given nucleus.
- The last value determined from the "Compute Defaults" button is the "Total Points" of the sample schedule.
- The total points are set to ~ 37% of the maximum increment for 1D schedules, 15% for 2D schedules, and 6% for 3D schedules.
- These percentages are conservative and smaller values can often be used.
- The "Compute Defaults" button is only provided to give estimates of values I feel are reasonable. The user can choose to change any of these values after the "Compute Defaults" button is pressed or simply enter their own values directly to create a sample schedule and not even use the "Compute Defaults" button.
- Maximum Increment
(top)
- This value sets the maximum increment number for each of the dimensions. For example if the maximum increment value for T1 and T2 were set to 96 and 133 then 96 and 133 would be the maximum t1 and t2 increments included in the sample schedule.
- The mouse-over information button provides the evolution time that the maximum increment corresponds to based on the sweep widths [evolution time = (1/sweep_width)*maximum_increment].
- The last point in all sample schedules is set to the maximum increment in all dimensions. This point in the experiment would correspond to a very weak signal, assuming that the experiment is not constant time. While it may seem silly to include a point with very weak signal it is often advantageous to be able to estimate the noise in the spectrum when processing with Maximum Entropy reconstruction so it is added to the sample schedule by default. The last line of the sample schedule can be deleted if you choose not to include it.
- Decay Rate
(top)
- Sample schedules are created randomly, but with a skewed distribution towards early increment time points where the NMR signal is likely to be stronger. The decay rate sets the "skewness" of the distribution. A larger decay rate will ensure that a higher proportion of the picked points are from early increments while a small decay rate will pick a distribution which is spread out more evenly across the sample schedule.
- The information mouse-over shows the linewidth for an NMR signal with a T2 value equal to the decay rate. It also shows the ratio between the decay rate linewidth and the expected linewidth based on the molecular weight (information shown in the nucleus information mouse over).
- By default the decay rate is set to 1.26X that of the expected R2 rate. This value is based on our prior experience as giving reasonable sample schedules, but can be edited by the user if desired.
- Decay Rate Force Checkbox (top)
- This checkbox forces the decay rate to be exactly what is selected when oversampling is set to greater than 1X.
- This button is ignored if oversampling is set to 1X.
- When oversampling is turned on (set to > 1X) the sweep widths are multiplied by the oversampling value, as are the maximum increment numbers. This is done internal to the program and is not reflected in the ScheduleTool window. The sample schedule is then calculated on this oversampled grid. If the decay rate is left the same as a non oversampled grid the schedule will be skewed significantly towards the early increment sample times.
- To eliminate this when oversampling is set to greater than 1X, and the Force checkbox is left unchecked, the program will first calculate a sample schedule with no oversampling and analyze the sample schedule to determine the average and median increment times. New sample schedules are then created with different decay rates on the oversampled grid until average and median increment times for each of the dimensions are achieved. Thus the oversampled sample schedule will have a similar distribution, relative to increment times, to a sample schedule without oversampling
- Force Initial Points
(top)
- Force initial points sets the number of points at the beginning of each dimension which are forced to be included into the sample schedule.
- The earliest increments have the greatest amount of signal and it is often desirable to make sure that these points are not skipped by random chance.
- The forced initial points does not add to the total number of points, they are included within it. If the total points is less than the product of all the force initial points, total points is recalculated to a suitable number. However, this is simply a uniform sample schedule and thus not useful.
- By default Force Initial Points cannot be set lower than 1. This means that the first point will always be selected.
- J-Coupling
(top)
- If your experiment is a J-modulation experiment, certain increment values will be nulled. By selecting SIN or COS modulation and providing the J-coupling values these null areas can be set to be less probable of being selected into the sample schedule.
- J-Coupling Value
(top)
- Oversampling
(top)
- When collecting indirect dimensions of NMR data that will be processed with the discrete Fourier transform it is desirable to set the sweep width to the minimum value needed to not alias signals. In fact the sweep width is often set even smaller to purposely fold signals in situations where the folded signals will not overlap with other non-folded signals. Setting the sweep width to these minimal values ensures that the resolution is as high as possible for a given number of collected time increments.
- By using Non-uniform sampling there is no reason that sampling must occur on the same time increment grid as a uniformly sampled experiment. Essentially the time increments can be set to any arbitrary value. However, for practical reasons it is often desirable to restrict the allowable time increments to be on some grid, although that grid can have a smaller time increment than a uniformly sample experiment and hence a larger sweep width.
- One easy way to accomplish this is to sample from an oversampled grid in the same manner than the acquisition dimension performs digital oversampling.
- To do this the sweep widths and maximum increment numbers are simply multiplied by an oversampling value such as 4X.
- The last point still corresponds to the same time value as a non-oversampled grid as the time increment between each point is smaller in proportion to the amount of oversampling.
- When randomly selecting a sample schedule from an oversampled grid with a skewed distribution based on an exponentially decaying function many more early time increments will be selected as compared to a non-oversampled sample schedule with the same decay rate. This is simply due to the greater number of points that may be sampled.
- For example for 2 indirect non-uniformly sampled dimensions with 4X oversampling there will be 16X as many possible points to choose from.
- To resolve this issue the sample scheduler will automatically try various decay rates when oversampling is turned on until it finds a sample schedule with a similar time distribution to one created without oversampling.
- The Force checkbox overrides this feature and uses the decay rate entered in the program no matter if oversampling is turned on or off.
- When processing oversampled collected spectra the intermediate file sizes can become quite large as the number of points in each dimension must be increased by the oversampling value. However, after processing, the central region of interest can be kept and all other regions deleted so that the final file size for analysis remains the same as if the data was collected without oversampling.
- Total Points
(top)
- Total points to be picked in the sample schedule. The percentage of possible points is shown. If oversampling is turned to greater than 1X the total number of points in the oversampled grid is also shown.
- Note that the number is the size of the oversampled grid, not the number of points collected when oversampling is turned on. That number is always equal to total points.
- Create Schedule
(top)
- Button that creates the sample schedule and opens the sample schedule window.
- When creating a sample schedule a progress bar is shown so that it can be estimated how long it will take to create.
- For non-oversampled schedules this time is often negligible. However it can take some time for a large 3D sample schedule to be created.
- For large 2D and 3D sample schedules created with oversampling the time can increase significantly. This is due to the much larger number of points in the oversampled grid and the fact that when oversampling is turned on and the Force checkbox is off that the program may need to calculate several sample schedules with different decay rates to achieve the desired distribution of points.
- Abort
(top)
- The abort button appears when a sample schedule is being created to allow the user to abort the process if it is taking too long.
- Help
(top)
- Button that opens a web browser pointing to this document.
SAMPLE SCHEDULE WINDOW (top)
The sample schedule window opens automatically after creating a sample schedule when running in GUI mode. The window is composed of five parts; A schedule window, a FFT window, a Stats window, and File and Data Menu bars.
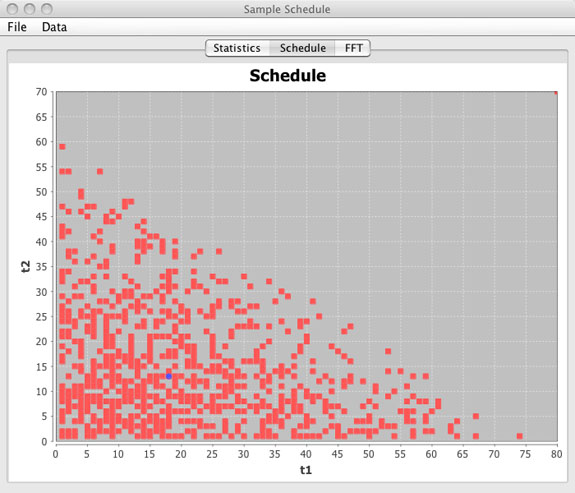
Schedule Window
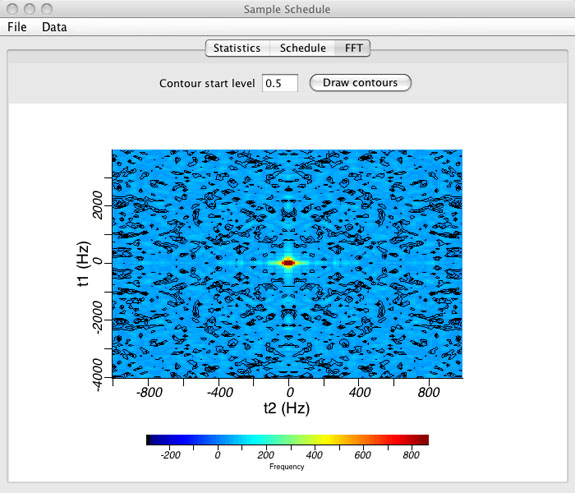
FFT or Point Spread Function Window
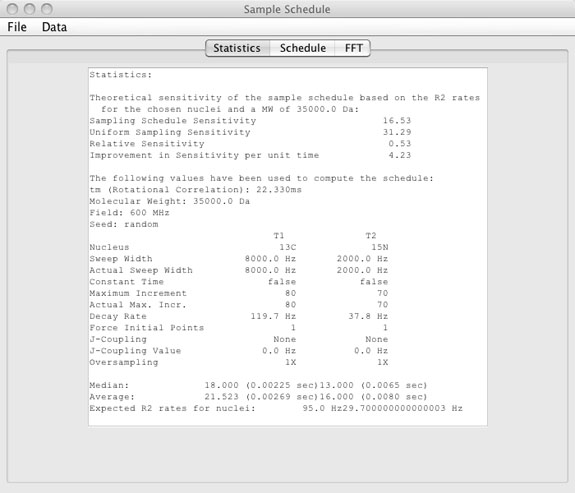
Statistics Window
- Graphical view of the sample schedule.
(top)
- This is a graphical representation of the sample schedule with points being collected shown as red squares. Sample points not in the sample schedule are blank. The blue circle represents the median increment and may or may not actually be included in the sample schedule.
- For 1D and 2D schedules the entire schedule is displayed as a single figure. For 3D schedules only the t1 / t2 dimensions are shown and a slider bar is included to view the t1 / t2 schedule planes for any given t3 value. Averages are shown per t1 / t2 slice.
- Using the mouse any region can be zoomed. The right mouse button may be used to zoom back out by selecting Auto Range - Both Axis.
- Graphical view of the point spread function (PSF).
(top)
- The point spread function is displayed by selecting the FFT tab. The PSF is shown with contours and as an intensity plot.
- The contour start level can be changed manually. Enter zero to turn contours off.
- The PSF is calculated by performing a discrete Fourier transform of the sample schedule where all points that are included in the sample schedule are set to a value of "one" and all points not included in the sample schedule are set to a value of "zero".
- When collecting NUS data there are often sampling artifacts that appear in the spectrum. These artifacts arise because each signal in the spectrum is convolved with the PSF. These artifacts are normally weak and can be ignored, but in cases where the PSF gives large intensities outside the central region (0 Hz) can lead to significant artifacts in the final spectrum. Therefore a sample schedule that minimizes these large amplitudes in the PSF will tend to give spectra with fewer intense sampling artifacts. In addition, using a processing method such as maximum entropy reconstruction (MaxEnt) is beneficial in that the sampling artifacts are deconvolved from the final spectrum to some degree without affecting any "true" signals (signals that are not arising due sampling artifacts).
- In general the larger the percentage of points and the more random the sample schedule the better the PSF will look. Sample schedules with any type of pattern such as those used in back projection will lead to the most significant sampling artifacts. In addition the central component is broadened slightly as the percentage of kept points in the sample schedule decreases. This will cause a slight broadening of the peaks in the final spectrum. As this is slight, and NUS generally affords much better resolution to begin with, this slight broadening is often ignored.
- Note however that MaxEnt can easily deconvolved this added linewidth from the spectrum and can even deconvolve some of the true linewidth of the peaks without causing deleterious effects to the spectrum (essentially narrowing all the peaks) as long as it is applied in a conservative manner. Conservative means that no more than 1/2 of the natural linewidth is deconvolved and the size of the final spectrum is large enough to eliminate truncation artifacts that may arise with the decrease in linewidth.
- Statistics window
(top)
- The statistics window shows values for:
- Sample schedule sensitivity
- This value is simply the sum of the exponential decay values for the given T2 based on the molecular weight, sweep widths, and nucleus choice for all points that are included in the sample schedule.
- Uniform sampling sensitivity
- This value is the same as the sampling schedule sensitivity except that it is the sum over all points up to the maximum increment and shows what the sensitivity would be if all the points were collected.
- Note: This value will likely be higher than the sampling schedule sensitivity. However, the experimental time would be much greater to achieve this sensitivity. Generally the sensitivity per unit time is greater for NUS sample schedules than uniform sampling.
- Relative sensitivity
- Simply the ratio of sampling schedule sensitivity and uniform sampling sensitivity.
- Improvement in sensitivity per unit time
- Simply the relative sensitivity normalized for experiment time.
- Rotational correlation time
(tm)
- The tm is based on the MW and is based on experimental relaxation data from the literature and not a theoretical value. The tm is slightly higher than the theoretical value.
- Parameters used to calculate the sample schedule.
- These values are for recording the parameters used to create the sample schedule. Note that sweep width, maximum increment, and decay rate have the values chosen and the actual values used to calculate the sample schedule. For sample schedules created without oversampling these values are identical, but may be different for sample schedules created with oversampling depending on wheter the force decay rate checkbox was selected.
- Median
- The median increment in each of the dimensions along with the evolution time that corresponds to that increment.
- Average
- The average increment number in each of the dimensions along with the evolution time that corresponds to that increment.
- R2
- The R2 relaxation rate expected based on the molecular weight and the nucleus choice.
- File Menu
(top)
- Save Varian
- Saves the sample schedule in a format suitable for using Orekhov / Hoch method of collecting NUS data from BioPack sequences. Also saves a second file suitable for processing the data using the Rowland NMR Toolkit.
- The difference is that the Varian file starts with 0 as the first increment while the toolkit file starts from 1.
- Save Bruker
- Saves the sample schedule in a format suitable for using the Wagner method of collecting NUS data on Bruker instruments.
- Save PSF-RNMRTK script
- Saves a Rowland NMR Toolkit processing script that will synthetically generate a data set and produce a point spread function of the sample schedule. The values in the PSF are identical between this tool and the toolkit.
- Save FFT result
- Saves a text file with the result of the point spread function. Both the real and imaginary components are saved in separate columns.
- Save peaks
- The sample schedule tool has a rudimentary peak picker. The save peaks command saves a text file with the output from the peak picker.
- Output is sorted from largest signal (the central component) to the smallest picked peak.
- The peak picker picks the peaks automatically upon the creation of the sample schedule so no peak picking is necessary to save the peak pick results.
- See command line
- The ScheduleTool can be run in GUI mode and from a command line. The "See command line" option allows the user to see in GUI mode the command line that was used to build the sample schedule. It is hoped that this can be used as a tutorial on how to build sample schedules from the command line.
- Print
- Prints the sample schedule window.
- Version
- Opens a window showing the current version number.
- Close
- Closes the Sample Schedule Window.
- NOTE: This does not close the ScheduleTool, just the Sample Schedule window. It is a known bug that having multiple sample schedule windows open at the same time may lead to problems. For instance if multiple sample schedule windows are open and the save peaks command is issued there is no guarantee that the correct PSF will be analyzed. I strongly urge users to only have a single sample schedule window open at a single time.
- Data Menu
(top)
- FFT data
- Shows the intensity for both real and imaginary points from the point spread function. This display only shows 1000 data points at a time. The user can choose the first point to view.
- Under the File Menu the result of the FFT can be saved. This will save all the data points and not just 1000 at a time.
- Peaks
- Displays a list of peaks (from a default peak picker) in a window. Peaks are shown in decreasing intensity with the central component at 0 Hz at the top.
DESCRIPTION OF THE COMMAND LINE (top)
The command line option for running the ScheduleTool can be used in two ways. The first is to simply create a sample schedule direct from the command line without any graphical interface. The second is to pass arguments to the ScheduleTool so that when the GUI of the ScheduleTool opens the passed arguments are entered by default rather than the pre-programmed default values.
Whenever the ScheduleTool opens the first thing that is done is information for the number of dimensions, the molecular weight, the sweep widths, field strength, and the nucleus types are refactored to determine suitable parameters for maximum increment, decay rates, and total points. Because of this the number of dimensions, molecular weight, sweep widths, field, nucleus choices, and the sample schedule filename are required when being used from the command line alone. For the GUI mode default values are used for the initial refactoring and after the user defines these parameters the compute defaults button may be pressed to update the parameters again.
Optional values for parameters to generate the sample schedule can also be entered from the command line. When these optional parameters are entered from the command line the program first ignores these values. The required entries (sweep widths, field, nucleus choices, molecular weight, and the number of dimensions) are processed to determine suitable values to generate the sample schedule. Then the optional parameters that were entered from the command line are used to replace the default values and the sample schedule is created.
In order to generate a sample schedule the molecular weight, field, and nucleus choices are not strictly needed. However, the way the ScheduleTool is written these values still need to be entered. Maybe in a future version I will check to see if all parameters are entered then the requirement for the molecular weight, field, and nucleus choices will be removed. For now you must enter these values.
In GUI mode once a sample schedule has been created the user may see the command line that would create the same sample schedule by going to the File Menu and click on See Command Line. It is hoped that this feature would act as a tutorial on how to use the command line.
NOTE: The required paramters ndim, molWeight, field, swtN, and nucltN must be entered before any non required parameters for the command line parser to work properly. Also, the command line parser is not tolerant of typos.
COMMAND LINE HELP (top)
Usage:
[--help] to print this
[--gui] to run in graphical mode
--- Required for GUIless mode ----------------------------------------
[--ndim {1,2,3}] Number of non-uniform dimensions (default: 2)
[--molWeight {x.x}]
[--field {500,600,700,800,900}]
[--swtN {x.x}] Sweep width tN
[--nucltN {1H-homo, 1H-13C, 1H-15N, 13C, 15N, 15N-TROSY}] Nucleus tN
[--sched {filename}] Schedule output file (for GUIless mode)
[{--bruker, --varian}] Schedule will be generated in Bruker/Varian format (one at a time, default: Varian)
--- Optional ----------------------------------------------------------
[--constTimetN {true, false}] Constant time tN
[--maxIncrtN {x.x}] Maximum increment tN
[--decayRatetN {x.x}] Decay Rate tN
[--forceFirsttN {x}] Force first x points tN
[--jcoupltN {None,sin,cos}] J-Coupling tN
[--jcoupleFreqtN {x.x}] J-Coupling frequency tN
[--oversampltN {1X, 2X,4X, 8X}] Oversampling tN
[--totalPoints {x}] Force total points
[--seed {x}] The seed for the random number generator
[--path {path}] The location where all the output files will be stored (default: current directory)
[--fftdata {filename}] Where to output the result of the fft function
[--peaks {filename}] Where to output the peaks in the fft'ed data
[--rnmrtk {filename}] Where to output the PSF script for RNMRTK
Example
java -jar ScheduleTool.jar --ndim 2 --molWeight 30000 --field 600 --nuclt1 15N --nuclt2 13C --swt1 2000 --swt2 8000 --path ./ --sched sched.dat